The Nylonase Story: How Unusual Is That?
Ann Gauger
Editor’s note: Nylon is a modern synthetic product used in the manufacturing, most familiarly, of ladies’ stockings but also a range of other goods, from rope to parachutes to auto tires. Nylonase is a popular evolutionary icon, brandished by theistic evolutionist Dennis Venema among others. In a series of three posts, of which this is the second, Discovery Institute biologist Ann Gauger takes a closer look.
In an article yesterday, “The Nylonase Story: When Imagination and Facts Collide,” I described how some biologists claim that the enzyme nylonase demonstrates that it is easy to get new functional proteins. It has been proposed that nylonase is the result of a frameshift mutation that produced an entirely new coding sequence from an alternate reading frame. I showed why such a claim is false. Now I will explain what that means and something about the unusual properties of the nylB gene that caught molecular geneticist and evolutionary biologist Susumu Ohno’s attention.
What are alternate reading frames? To answer that question, I first need to provide some background information. I will begin by defining some terms I used in yesterday’s post. DNA is composed of two anti-parallel strands of nucleotides. The order of the nucleotides in each strand is what specifies the information the DNA carries. The two strands, called the sense and antisense strands, run in opposite directions. Even though their sequences are complementary, with A always paired with T, and C with G, each strand carries different potential information.
ATG GCA TGC ACC GGC ATT AG → sense
TAC CGT ACG TGG CCG TAA TC ← antisense
Before the information in DNA can be used, it must be copied into what we call messenger RNA. The sequence of one strand of DNA, usually the sense strand, is copied using the same base complementarity: G pairs with C, and A with U (U is used in place of T in RNA). We call that copying transcription. The message that has been transcribed from the DNA into that sequence of RNA is now ready to be translated into protein.
Notice the language of information shot throughout these processes. The names for these processes were given by men fully committed to a naturalistic worldview, men such as Francis Crick and Sydney Brenner. Indeed, they were materialists one and all. Yet they saw the parallels between these processes and the human manipulation of text (language) or code (another form of language). The genetic code is the framework that determines the relationship between groups of nucleotides (codons), and the amino acids they specify. The code specifies how to translate the messenger RNA that has been copied or transcribed from the DNA, so that it can be translated into a new language, the language of proteins. Below is an illustration of the standard genetic code (source here, used with permission):
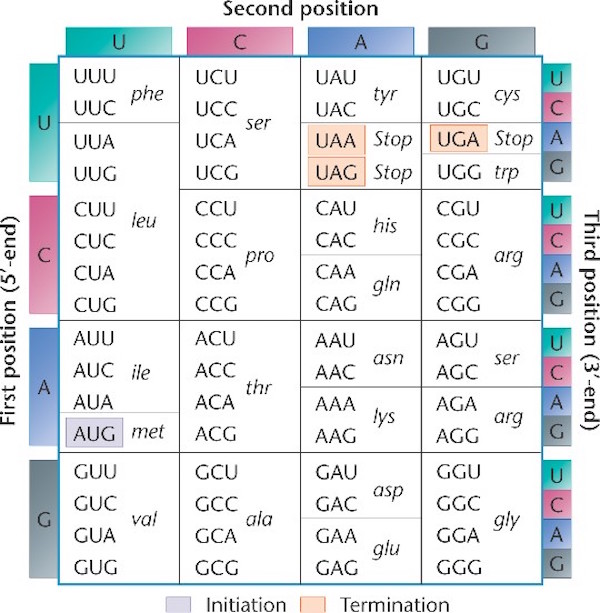
Notice that the information in DNA is read in groups of three nucleotides (each group is called a codon), and each codon specifies a particular amino acid. Sometimes more than one codon can specify the same amino acid. For example in the top left corner, the table shows that UUU and UUC both specify the amino acid phenylalanine.
The nature of the code is such that it matters where the first codon begins — the first codon to be read establishes the codon groupings going forward. In the table above the “start” codon is AUG (it also specifies the amino acid methionine). The sequence of codons is “read” by a cellular machine called the ribosome, which starts reading the RNA message at AUG, and then proceeds three nucleotides at a time to translate the message into amino acids. In the sequence below, for example, the first codon to be read would be AUG and that codon determines the frame in which of all the other codons are read.
AUG GCA UGC ACC GGC AUU AGU
Now here’s where it gets interesting. Potentially, DNA can be grouped into different codons, or frames, depending on where the ribosome starts reading. See below for an illustration. For example, the sequence could potentially be read with the groupings shown in frame one (ATG GCA etc.) or frame two (TGG CAT etc., if a proper ATG exists somewhere upstream), leading ultimately to a different amino acid sequences for each. In fact there are six possible ways to group the DNA into codons — three frames on the sense strand going left to right (labeled 1-3), and three frames on the antisense strand (labeled 4-6), going right to left. Below I have laid out the six possible frames for the sequence we began with, but with the alternate frames staggered, and the alternate codons separated by spaces. Notice the sequence stays the same — the only thing that changes from frame to frame is how the nucleotides are grouped. It’s the same sequence, but it could be read and translated differently in each frame. This is because each codon specifies a particular amino acid. Thus, each frame results in a completely different string of amino acids.
frame 1 ATG GCA TGC ACC GGC ATT AG
frame 2 TGG CAT GCA CCG GCA TTA G
frame 3 GGC ATG CAC CGG CAT TAG
frame 4 TAC CGT ACG TGG CCG TAA TC
frame 5 ACC GTA CGT GGC CGT AAT C
frame 6 CCG TAC GTG GCC GTA ATC
The codons TAA, TAG, and TGA are stop codons — they specify where the gene ends and protein translation stops. (For extra credit, can you find any ATG or stop codons in the above frames? They are there in both the forward and reverse direction. For more extra credit, can you use the code table to translate different frames, and demonstrate that each frame encodes a different protein?)
So when Venema and others say that nylonase arose by a frameshift mutation that produced a novel protein 392 amino acids long, they are claiming that a completely new coding sequence with frame-shifted codons could generate a functional protein. How likely is that? Not very, given the rarity of functional proteins in sequence space (see my first post). And, as I have already shown in my first post, such an unlikely hypothesis is unnecessary. The nylB gene appears to be the product of a simple gene duplication followed by two stepwise mutations to increase nylonase activity.
There is something special about the nylonase gene’s sequence though, something very odd. nylB has multiple large, overlapping (alternate) open frames that lack stop codons.
How hard is it to get a gene with multiple reading frames?
Let me explain. Roughly one in twenty codons are stop codons. A random DNA sequence will have stop codons about every sixty bases, and may or may not have a start codon. Usually the alternate frames of DNA sequences are interrupted by stop codons. Only the frame that actually specifies the correct gene will have no stop codons at all over a significant length. This system is actually very ingenious. The one frame that needs to be read and translated is identified by an ATG. The other frames will usually lack an ATG and/or will have several stop codons that interrupt their translation, thus preventing the cell from wasting energy on nonsense transcripts.
According to the nylonase story, as told by Ohno and Venema and numerous others, a new ATG start codon was formed by the insertion of a T between an A and G, thus creating a new start codon after the original ATG, which shifted the reading frame for that sequence to that specified by the new ATG, and creating a completely different coding sequence and thus a new protein. Let us grant that scenario for the sake of argument. Normally such a shift would produce a new coding sequence that would be interrupted by stop codons, so the newly frameshifted protein would be truncated. Thus the only reason this frameshift hypothesis for nylonase is even remotely possible is because the sequence coding for nylonase is most unusual, and contains not one, not two, but three open frames Although frameshift mutations are ordinarily considered to be quite disruptive, at least in this case the putative brand new protein sequence would not terminate early due to stop codons.
My point? The first step to getting a new functional protein of any length from a frameshift is to avoid stop codons. The odds of a random coding sequence having an open alternate frame, without stops, are poor. As a consequence, if a protein does have an open frame in addition to its coding sequence, it’s worth paying attention to. And it so happens that nylonase does have more than one open frame. The DNA sequence above illustrates the six frames, numbering them frames 1 through 6. Using that convention, frames 1 and 3 are read from the sense strand. Both have no stop codons over the length of the gene in the sense direction. Frame 4 on the antisense direction has no stop codons either. Frame 1 is the coding frame that specifies the nylonase protein, otherwise known as the open reading frame (ORF). It is defined by the presence of both a start and stop codon. The other two frames have no start codons or stop codons, so I’ll call them non-stop frames (NSFs). They are frames 3 and 4.
The probability of a DNA sequence with an ORF on the sense strand and 2 NSFs is very small. Just exactly how small are the chances of avoiding a stop codon in three out of six frames? We set out to determine that by performing a numerical simulation using pseudorandom numbers to generate sequences at various levels of GC content. (By we I mean that my husband, Patrick Achey, who is an actuary, did the programming work, while I determined the parameters.) We chose to vary the GC content because sequences with a higher GC content have fewer stop codons. Remember, a stop codon always has an A and a T (TAA, TAG, and TGA are the stop codons) so having a sequence with a lower percentage of AT content will reduce the frequency of stop codons. Conversely, higher GC content makes the chances of avoiding stop codons and getting longer ORFs much greater, thus also increasing the chances of NSFs. The genomes of bacteria vary in their GC content, from less than 20 percent to as much as 75 percent, though the reason why is not known. One species of Flavobacterium has a genome with about 32 percent GC and 2400 genes — the precise values varies with the strain. The plasmid on which nylB resides is very different. It has 65 percent GC content. The gene encoding nylonase has an even higher 70 percent GC content, which is near the observed bacterial maximum of 75 percent.
We chose to use a target ORF size of 900 nucleotides (or 300 amino acids) because it is an average size for a functional protein. Nylonase is 392 amino acids long; the small domain of beta lactamase, the enzyme my colleague Doug Axe studied, is about 150 amino acids long. The median length for an E. coli protein is 278 amino acids; for humans, the median length is 375.
As expected, the simulation showed that the higher the GC content, the greater the likelihood that ORFs that are 900+ nucleotides long exist. At 50 percent GC, the average ORF length we obtained was about 60 nucleotides; most ORFs terminate well before 900 nucleotides. Indeed, in our simulation only two out of a million random sequences made it to 900 nucleotides before encountering a stop codon. As a result, we could not determine the rarity of NSFs at 50 percent GC — we would probably have to run the simulation for more than a billion trials to get any significant number of NSFs at all.
Sequences at 60 percent GC gave 57 ORFs at least 900 nucleotides long out of a million trials, while sequences at 65 percent GC produced 404 out of a million, one of which also had an NSF.
NSFs were much more probable for sequences that were 70 percent GC, like nylB. In our simulation 3,021 out of a million trials were ORFs at least 900 nucleotides long. That’s a frequency of .3 percent. Of those 3,021 ORFs, 86 had 1 NSF, and none had 2 NSFs. We had to run 10 million trials at 70 percent GC to see any ORFs with 2 NSFs. From those 10 million randomly generated sequences, we obtained 28,603 ORFs; 903 had 1 NSF and only 9 had 2 NSFs.
Interestingly, at 80 percent GC we got a few sequences with 4 NSFs; but I don’t know of any bacterium with a GC content that high.
Our simulation shows that multiple NSFs are very rare. The probability that an ORF 900 nucleotides long with 70 percent GC content will have two NSFs is 9 out of 28,603, or 0.0003. If these figures are recast to include the total number of trials required to get an ORF of that length and GC content and with 2 NSFs, the probability would be 9 out of 10,000,000 trials.
A sequence like nylB is very rare. In fact, I suspect that for all cases where overlapping genes exist, in other words where alternate frames from the same sequence have the potential to code for different proteins, unusual sequence will necessarily be found. Likely it will be high in GC content. Could such rare sequences be accidental? I think that if we compare the expected number of alternate or overlapping NSFs per ORF, with the actual number we will find that there are more of these alternate open reading frames than would be predicted by chance.
From another study of overlapping genes:
Thus, bacterial genomes contain a larger number of long shadow ORFs [ORFs on alternate frames] than expected based on statistical analysis. Random mutational drift would have eliminated the signal long ago, if no selection pressures were stabilizing shadow ORFs. Deviations between the statistical model and bacterial genomes directly call for a functional explanation, since selection is the only force known to stabilize the depletion of stop codons. Most shadow genes have escaped discovery, as they are dismissed as false positives in most genome annotation programs. This is in sharp contrast to many embedded overlapping genes that have been discovered in bacteriophages. Since phages reside in a long term evolutionary equilibrium with the bacterial host genome, we suggest that overlooked shadow genes also exist in bacterial genomes.
Indeed, a study of the pOAD2 plasmid from which nylB came indicates that there are potentially many overlapping genes on that plasmid. nylB′, for example, a homologous gene on the same plasmid that differs by 47 amino acids from nylB, also has 2 NSFs. These unusual and unexpected features of DNA have consequences for how we think about the origin of information in DNA sequences, as I shall discuss in the next post.
Ann Gauger
Editor’s note: Nylon is a modern synthetic product used in the manufacturing, most familiarly, of ladies’ stockings but also a range of other goods, from rope to parachutes to auto tires. Nylonase is a popular evolutionary icon, brandished by theistic evolutionist Dennis Venema among others. In a series of three posts, of which this is the second, Discovery Institute biologist Ann Gauger takes a closer look.
In an article yesterday, “The Nylonase Story: When Imagination and Facts Collide,” I described how some biologists claim that the enzyme nylonase demonstrates that it is easy to get new functional proteins. It has been proposed that nylonase is the result of a frameshift mutation that produced an entirely new coding sequence from an alternate reading frame. I showed why such a claim is false. Now I will explain what that means and something about the unusual properties of the nylB gene that caught molecular geneticist and evolutionary biologist Susumu Ohno’s attention.
What are alternate reading frames? To answer that question, I first need to provide some background information. I will begin by defining some terms I used in yesterday’s post. DNA is composed of two anti-parallel strands of nucleotides. The order of the nucleotides in each strand is what specifies the information the DNA carries. The two strands, called the sense and antisense strands, run in opposite directions. Even though their sequences are complementary, with A always paired with T, and C with G, each strand carries different potential information.
ATG GCA TGC ACC GGC ATT AG → sense
TAC CGT ACG TGG CCG TAA TC ← antisense
Before the information in DNA can be used, it must be copied into what we call messenger RNA. The sequence of one strand of DNA, usually the sense strand, is copied using the same base complementarity: G pairs with C, and A with U (U is used in place of T in RNA). We call that copying transcription. The message that has been transcribed from the DNA into that sequence of RNA is now ready to be translated into protein.
Notice the language of information shot throughout these processes. The names for these processes were given by men fully committed to a naturalistic worldview, men such as Francis Crick and Sydney Brenner. Indeed, they were materialists one and all. Yet they saw the parallels between these processes and the human manipulation of text (language) or code (another form of language). The genetic code is the framework that determines the relationship between groups of nucleotides (codons), and the amino acids they specify. The code specifies how to translate the messenger RNA that has been copied or transcribed from the DNA, so that it can be translated into a new language, the language of proteins. Below is an illustration of the standard genetic code (source here, used with permission):
Notice that the information in DNA is read in groups of three nucleotides (each group is called a codon), and each codon specifies a particular amino acid. Sometimes more than one codon can specify the same amino acid. For example in the top left corner, the table shows that UUU and UUC both specify the amino acid phenylalanine.
The nature of the code is such that it matters where the first codon begins — the first codon to be read establishes the codon groupings going forward. In the table above the “start” codon is AUG (it also specifies the amino acid methionine). The sequence of codons is “read” by a cellular machine called the ribosome, which starts reading the RNA message at AUG, and then proceeds three nucleotides at a time to translate the message into amino acids. In the sequence below, for example, the first codon to be read would be AUG and that codon determines the frame in which of all the other codons are read.
AUG GCA UGC ACC GGC AUU AGU
Now here’s where it gets interesting. Potentially, DNA can be grouped into different codons, or frames, depending on where the ribosome starts reading. See below for an illustration. For example, the sequence could potentially be read with the groupings shown in frame one (ATG GCA etc.) or frame two (TGG CAT etc., if a proper ATG exists somewhere upstream), leading ultimately to a different amino acid sequences for each. In fact there are six possible ways to group the DNA into codons — three frames on the sense strand going left to right (labeled 1-3), and three frames on the antisense strand (labeled 4-6), going right to left. Below I have laid out the six possible frames for the sequence we began with, but with the alternate frames staggered, and the alternate codons separated by spaces. Notice the sequence stays the same — the only thing that changes from frame to frame is how the nucleotides are grouped. It’s the same sequence, but it could be read and translated differently in each frame. This is because each codon specifies a particular amino acid. Thus, each frame results in a completely different string of amino acids.
frame 1 ATG GCA TGC ACC GGC ATT AG
frame 2 TGG CAT GCA CCG GCA TTA G
frame 3 GGC ATG CAC CGG CAT TAG
frame 4 TAC CGT ACG TGG CCG TAA TC
frame 5 ACC GTA CGT GGC CGT AAT C
frame 6 CCG TAC GTG GCC GTA ATC
The codons TAA, TAG, and TGA are stop codons — they specify where the gene ends and protein translation stops. (For extra credit, can you find any ATG or stop codons in the above frames? They are there in both the forward and reverse direction. For more extra credit, can you use the code table to translate different frames, and demonstrate that each frame encodes a different protein?)
So when Venema and others say that nylonase arose by a frameshift mutation that produced a novel protein 392 amino acids long, they are claiming that a completely new coding sequence with frame-shifted codons could generate a functional protein. How likely is that? Not very, given the rarity of functional proteins in sequence space (see my first post). And, as I have already shown in my first post, such an unlikely hypothesis is unnecessary. The nylB gene appears to be the product of a simple gene duplication followed by two stepwise mutations to increase nylonase activity.
There is something special about the nylonase gene’s sequence though, something very odd. nylB has multiple large, overlapping (alternate) open frames that lack stop codons.
How hard is it to get a gene with multiple reading frames?
Let me explain. Roughly one in twenty codons are stop codons. A random DNA sequence will have stop codons about every sixty bases, and may or may not have a start codon. Usually the alternate frames of DNA sequences are interrupted by stop codons. Only the frame that actually specifies the correct gene will have no stop codons at all over a significant length. This system is actually very ingenious. The one frame that needs to be read and translated is identified by an ATG. The other frames will usually lack an ATG and/or will have several stop codons that interrupt their translation, thus preventing the cell from wasting energy on nonsense transcripts.
According to the nylonase story, as told by Ohno and Venema and numerous others, a new ATG start codon was formed by the insertion of a T between an A and G, thus creating a new start codon after the original ATG, which shifted the reading frame for that sequence to that specified by the new ATG, and creating a completely different coding sequence and thus a new protein. Let us grant that scenario for the sake of argument. Normally such a shift would produce a new coding sequence that would be interrupted by stop codons, so the newly frameshifted protein would be truncated. Thus the only reason this frameshift hypothesis for nylonase is even remotely possible is because the sequence coding for nylonase is most unusual, and contains not one, not two, but three open frames Although frameshift mutations are ordinarily considered to be quite disruptive, at least in this case the putative brand new protein sequence would not terminate early due to stop codons.
My point? The first step to getting a new functional protein of any length from a frameshift is to avoid stop codons. The odds of a random coding sequence having an open alternate frame, without stops, are poor. As a consequence, if a protein does have an open frame in addition to its coding sequence, it’s worth paying attention to. And it so happens that nylonase does have more than one open frame. The DNA sequence above illustrates the six frames, numbering them frames 1 through 6. Using that convention, frames 1 and 3 are read from the sense strand. Both have no stop codons over the length of the gene in the sense direction. Frame 4 on the antisense direction has no stop codons either. Frame 1 is the coding frame that specifies the nylonase protein, otherwise known as the open reading frame (ORF). It is defined by the presence of both a start and stop codon. The other two frames have no start codons or stop codons, so I’ll call them non-stop frames (NSFs). They are frames 3 and 4.
The probability of a DNA sequence with an ORF on the sense strand and 2 NSFs is very small. Just exactly how small are the chances of avoiding a stop codon in three out of six frames? We set out to determine that by performing a numerical simulation using pseudorandom numbers to generate sequences at various levels of GC content. (By we I mean that my husband, Patrick Achey, who is an actuary, did the programming work, while I determined the parameters.) We chose to vary the GC content because sequences with a higher GC content have fewer stop codons. Remember, a stop codon always has an A and a T (TAA, TAG, and TGA are the stop codons) so having a sequence with a lower percentage of AT content will reduce the frequency of stop codons. Conversely, higher GC content makes the chances of avoiding stop codons and getting longer ORFs much greater, thus also increasing the chances of NSFs. The genomes of bacteria vary in their GC content, from less than 20 percent to as much as 75 percent, though the reason why is not known. One species of Flavobacterium has a genome with about 32 percent GC and 2400 genes — the precise values varies with the strain. The plasmid on which nylB resides is very different. It has 65 percent GC content. The gene encoding nylonase has an even higher 70 percent GC content, which is near the observed bacterial maximum of 75 percent.
We chose to use a target ORF size of 900 nucleotides (or 300 amino acids) because it is an average size for a functional protein. Nylonase is 392 amino acids long; the small domain of beta lactamase, the enzyme my colleague Doug Axe studied, is about 150 amino acids long. The median length for an E. coli protein is 278 amino acids; for humans, the median length is 375.
As expected, the simulation showed that the higher the GC content, the greater the likelihood that ORFs that are 900+ nucleotides long exist. At 50 percent GC, the average ORF length we obtained was about 60 nucleotides; most ORFs terminate well before 900 nucleotides. Indeed, in our simulation only two out of a million random sequences made it to 900 nucleotides before encountering a stop codon. As a result, we could not determine the rarity of NSFs at 50 percent GC — we would probably have to run the simulation for more than a billion trials to get any significant number of NSFs at all.
Sequences at 60 percent GC gave 57 ORFs at least 900 nucleotides long out of a million trials, while sequences at 65 percent GC produced 404 out of a million, one of which also had an NSF.
NSFs were much more probable for sequences that were 70 percent GC, like nylB. In our simulation 3,021 out of a million trials were ORFs at least 900 nucleotides long. That’s a frequency of .3 percent. Of those 3,021 ORFs, 86 had 1 NSF, and none had 2 NSFs. We had to run 10 million trials at 70 percent GC to see any ORFs with 2 NSFs. From those 10 million randomly generated sequences, we obtained 28,603 ORFs; 903 had 1 NSF and only 9 had 2 NSFs.
Interestingly, at 80 percent GC we got a few sequences with 4 NSFs; but I don’t know of any bacterium with a GC content that high.
Our simulation shows that multiple NSFs are very rare. The probability that an ORF 900 nucleotides long with 70 percent GC content will have two NSFs is 9 out of 28,603, or 0.0003. If these figures are recast to include the total number of trials required to get an ORF of that length and GC content and with 2 NSFs, the probability would be 9 out of 10,000,000 trials.
A sequence like nylB is very rare. In fact, I suspect that for all cases where overlapping genes exist, in other words where alternate frames from the same sequence have the potential to code for different proteins, unusual sequence will necessarily be found. Likely it will be high in GC content. Could such rare sequences be accidental? I think that if we compare the expected number of alternate or overlapping NSFs per ORF, with the actual number we will find that there are more of these alternate open reading frames than would be predicted by chance.
From another study of overlapping genes:
Thus, bacterial genomes contain a larger number of long shadow ORFs [ORFs on alternate frames] than expected based on statistical analysis. Random mutational drift would have eliminated the signal long ago, if no selection pressures were stabilizing shadow ORFs. Deviations between the statistical model and bacterial genomes directly call for a functional explanation, since selection is the only force known to stabilize the depletion of stop codons. Most shadow genes have escaped discovery, as they are dismissed as false positives in most genome annotation programs. This is in sharp contrast to many embedded overlapping genes that have been discovered in bacteriophages. Since phages reside in a long term evolutionary equilibrium with the bacterial host genome, we suggest that overlooked shadow genes also exist in bacterial genomes.
Indeed, a study of the pOAD2 plasmid from which nylB came indicates that there are potentially many overlapping genes on that plasmid. nylB′, for example, a homologous gene on the same plasmid that differs by 47 amino acids from nylB, also has 2 NSFs. These unusual and unexpected features of DNA have consequences for how we think about the origin of information in DNA sequences, as I shall discuss in the next post.